
In Press: Henry Miller Publishes First Author Paper in MDPI Cancers

 Researchers in the Greehey Children’s Cancer Research Institute recently published in MDPI Cancers as part of a special issue on Ewing sarcoma. Their study, titled “Reconstruction of Ewing Sarcoma Developmental Context from Mass-Scale Transcriptomics Reveals Characteristics of EWSR1-FLI1 Permissibility,” uses computational biology to answer fundamental questions about how Ewing sarcoma develops in patients.
Researchers in the Greehey Children’s Cancer Research Institute recently published in MDPI Cancers as part of a special issue on Ewing sarcoma. Their study, titled “Reconstruction of Ewing Sarcoma Developmental Context from Mass-Scale Transcriptomics Reveals Characteristics of EWSR1-FLI1 Permissibility,” uses computational biology to answer fundamental questions about how Ewing sarcoma develops in patients.
Ewing sarcoma is a pediatric cancer typically caused by the fusion of the EWSR1 to the FLI1 gene. The EWSR1-FLI1 fusion drives cells to divide uncontrollably and form a tumor. However, researchers do not know in which type of cell this occurs.
“To understand how Ewing sarcoma forms in patients, many groups have tried to express the EWSR1-FLI1 fusion gene in different cell types to see if they could make a Ewing sarcoma tumor,” said Henry Miller, a student in the Integrated Biomedical Sciences program and first author on the paper.
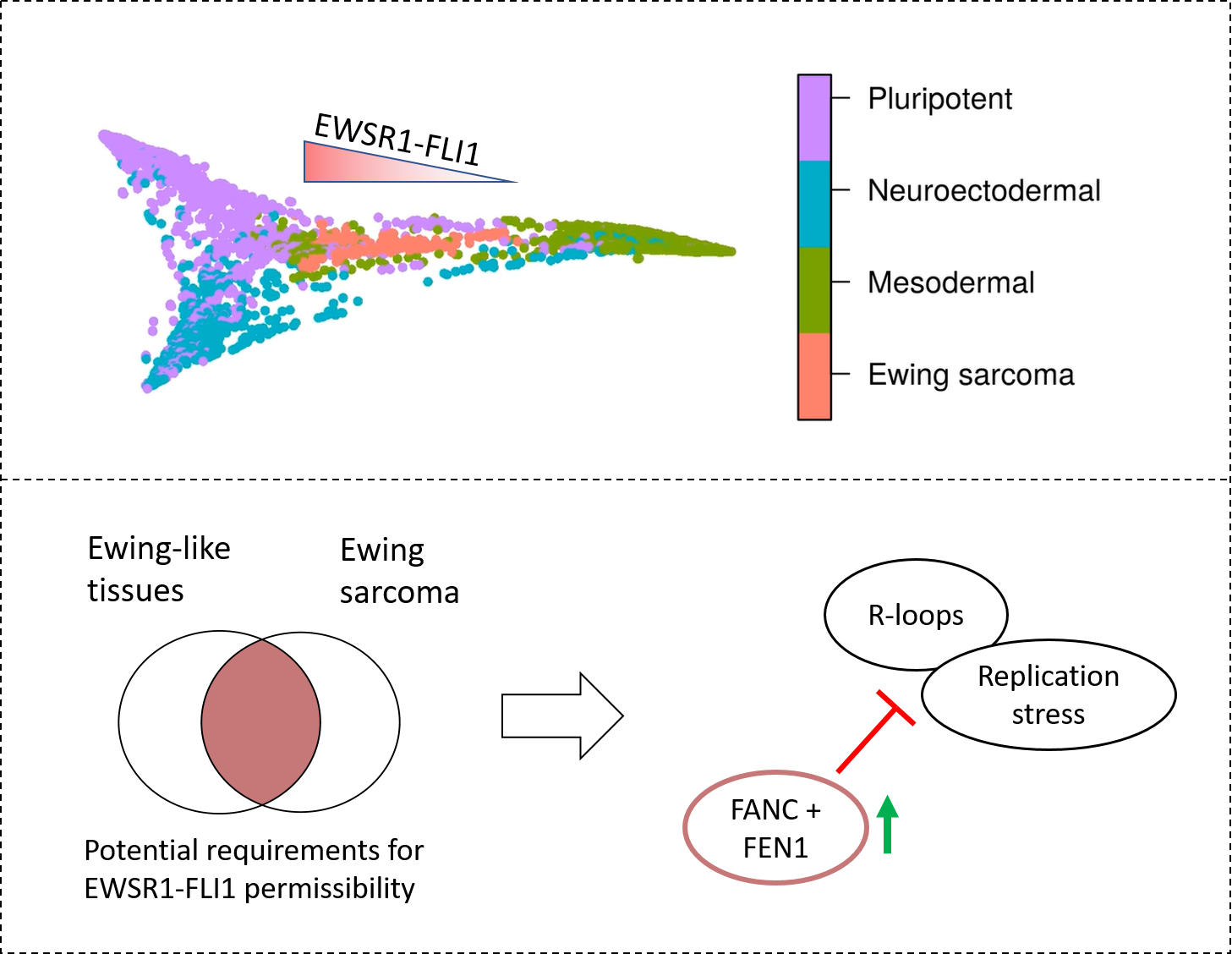
“In most cases EWSR1-FLI1 expression is toxic to the cell. This raised the question of which kinds of tissues can actually become Ewing sarcoma and why.”
According to Miller, “Our analysis showed that both Ewing sarcoma and the cells which could express EWSR1-FLI1 share the ability to mediate conflicts between transcription and replication. Because transcription-replication conflicts lead to genomic stress, we believe this means that Ewing sarcoma can only arise from cell types that can mediate these conflicts.”

More importantly, this research uncovers new drug targets for treating Ewing sarcoma patients.
“We demonstrated that Ewing sarcoma depends heavily upon these transcription-replication mediating factors,” explained Miller, “and we see them as promising targets for new clinical therapies.”
The work of Miller and colleagues offers a new perspective on research in which computational biology plays a central role.
“We didn’t know what we would find going into this project, but we saw an immense and untapped potential in publicly-available data to reveal something new about Ewing sarcoma.”
Miller and colleagues say that they re-analyzed over 40,000 publicly-available RNA-sequencing samples and that others could do the same even with limited computational resources.
“We got our data from the ARCHS4 database. They provide a user-friendly interface to find data and download it, or you can download every study at once like we did. With this database and some basic capability with a computing language like R or Python, it’s relatively easy to do an analysis like ours and discover something meaningful from it. To help facilitate this, we’ve made all of the R code used in our paper publicly available on github.” The link to the code base is https://github.com/millerh1/Ewing-sarcoma-paper-Miller-2020
With the increase in 2019-nCoV cases and social distancing policies, the freedom afforded by computational biology may be a viable alternative for researchers at UT Health San Antonio who are unable to return to the lab at present.
“Almost every research project can benefit from utilizing computational resources and publicly-available data,” says Miller, “so we hope that we can inspire others to explore the ARCHS4 database and see what they can uncover.”
In Press is a section in The Pipette Gazette that highlights publications by students. To read more In Press articles, click here.